

- Bone Health
- Immunology
- Hematology
- Respiratory
- Dermatology
- Diabetes
- Gastroenterology
- Neurology
- Oncology
- Ophthalmology
- Rare Disease
- Rheumatology
Analysis of FDA-Licensed Biosimilars: Time for a Paradigm Shift
Now is the time for the FDA to lead again in revising the biosimilar development guidance by eliminating all animal toxicology studies, and replacing them with larger-species pharmacokinetic (PK) studies, allowing the conduct of human PK studies using novel clinical protocols to combine the PK/pharmacodynamic/immunogenicity testing in a single study, and, where possible, avoid these studies if an in-silico approach can provide the confidence of pharmacologic similarity.
Sarfaraz Niazi, PhD
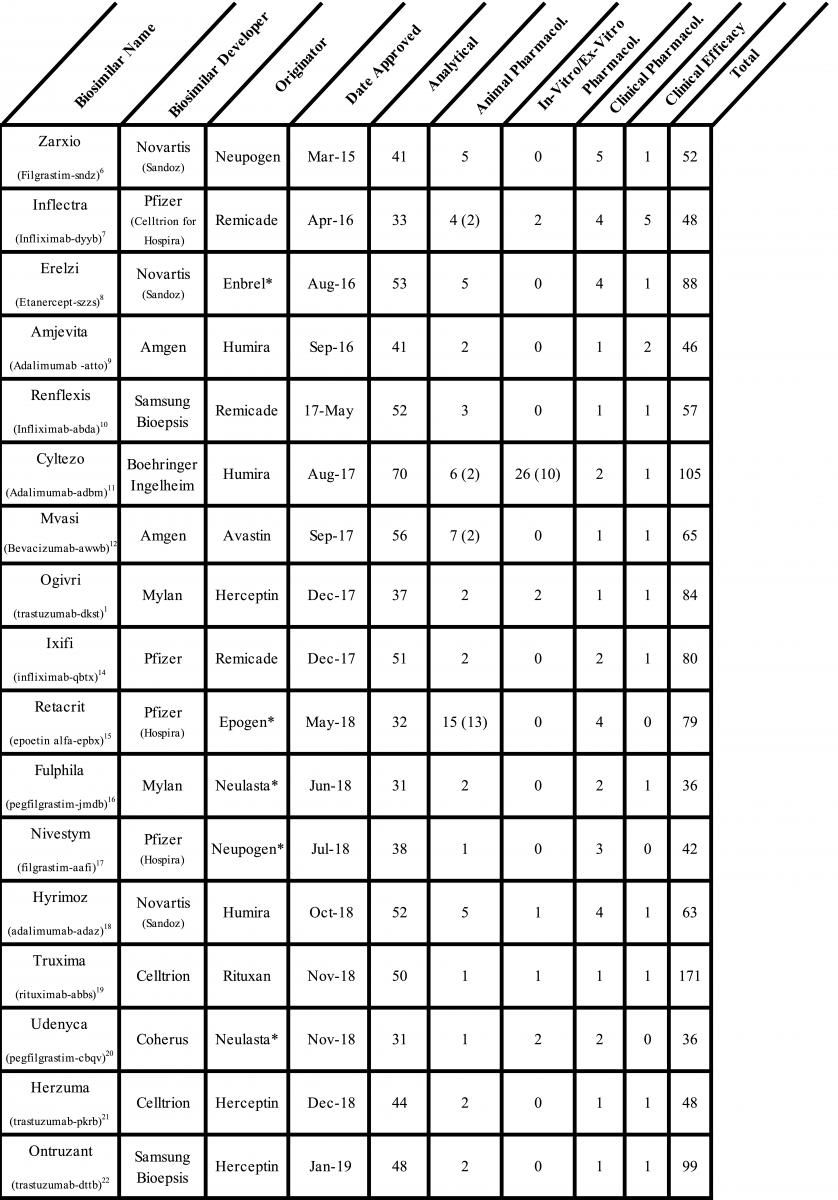
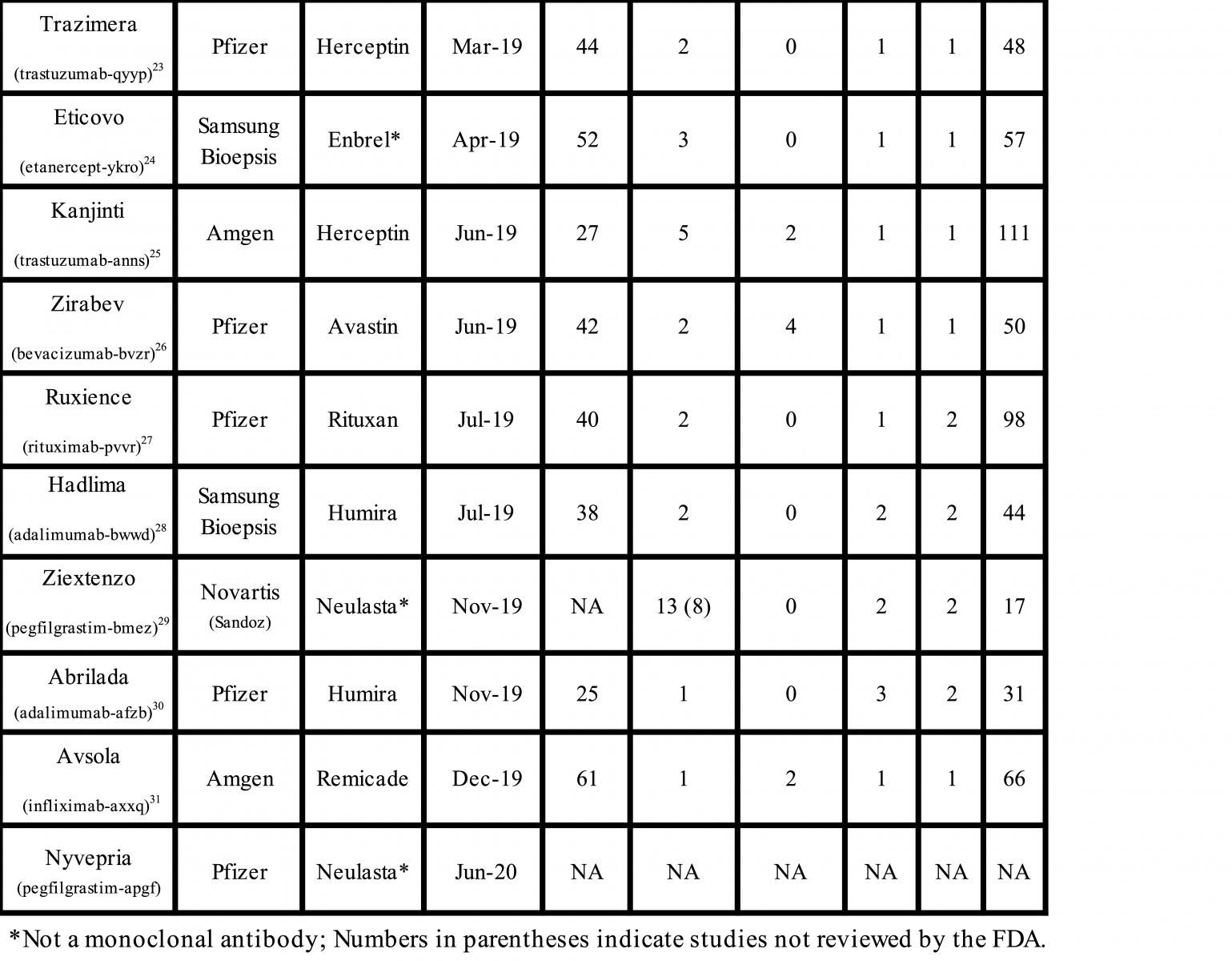
The FDA has approved 28 biosimilar products, 19 monoclonal antibodies, 6 cytokines, and 1 fusion protein since the first biosimilar was approved in 2015 (Table). A detailed analysis of the regulatory submissions that led to the approval of these products shows high diversity, frequent unnecessary testing, and reliance on studies that may not assure the safety and efficacy of biosimilars. The paradigm of stepwise development and evaluation suggested by the FDA has not worked either, and now we have sufficient data to indicate that a significant change in the biosimilars approval guidance is required to eliminate redundant testing and reduce the risk of approval of unsafe biosimilars.
The FDA got its charter to approve biosimilars in 2009.1 The FDA has issued 10 regulatory guidance documents2, and it redrafted 1 pivotal guidance on analytical testing3 after the author pointed out inconsistencies in the original.4 However, this fresh guidance still leaves out several key elements that are suggested in this column.
First, I will provide a broad summary of the scope and redundancies in the regulatory submissions from 8 companies among the 28 approvals. Not including the most recent applications for the pegfilgrastim biosimilar Nyvepria and adalumumab biosimilar Hulio (for which the FDA has not yet posted the Biologics License Application [BLA] details) there were over 1100 analytical similarity, 96 animal pharmacology, 42 in-vitro/ex-vitro pharmacology, 52 clinical pharmacology, and 32 clinical efficacy studies conducted. Here are the highlights of the compiled data:
- There was no consistency in the number of studies submitted for the same molecule: 5 adalimumab biosimilar applications included 31 to 105 studies; 5 trastuzumab biosimilar applications, 48 to 111 studies; and 4 infliximab biosimilar applications, 48 to 80 studies.
- Twenty-seven animal pharmacology studies were not reviewed by the FDA, which labeled them as them unnecessary or unwarranted.
- No animal pharmacology or in-vitro/ex-vitro studies failed.
- A few clinical pharmacology studies had to be repeated to meet acceptance criteria due to inappropriate choice of the study population. None failed.
- No clinical efficacy studies failed. There were no instances where the primary end points were not met. Additionally, post hoc analyses revealed no justification for invalidating the studies, nor were there additional scientific justifications for nulling these studies. In 2 cases, higher immunogenicity was overcome by making minor changes to the manufacturing process. No product was rejected based on a failed efficacy study.
In summary, all analytical similarity testing met the acceptance criteria, the animal pharmacology studies added little to the knowledge, all clinical pharmacology studies met the criteria, and even when there were differences in clinical efficacy studies, these were overcome by a discussion of data and marketing authorization was granted.
The FDA has long emphasized a stepwise approach to the development of biosimilars, and the stereotypical pyramid showing the importance of various studies has been circulated ad infinitum. The analytical similarity testing is the largest component and clinical efficacy, the smallest piece of the pyramid. However, the developers, all of them being large pharmaceutical companies with 1 exception, decided to conduct extensive, costly clinical efficacy testing; Coherus BioSciences, the exception, received approval without submitting a clinical efficacy study.
There are several fundamental arguments against the use of clinical efficacy testing to establish biosimilarity. First, no product is allowed to move forward to the clinical efficacy stage unless it meets analytical similarity and animal toxicology criteria. The FDA states that if there remains any “residual uncertainty,” additional testing may be required. Does this mean that the clinical efficacy testing is needed for the FDA to resolve any residual uncertainty? I could not find any such indication in any of the Biologics License Application documents that are in the public domain. Were the clinical efficacy studies conducted to support marketing plans or to support biosimilarity? None of the efficacy studies resulted in any rejection, which suggests that if a product meets the requirements up to the clinical pharmacology stage, it is not likely to fail in clinical efficacy testing.
There are additional reasons why clinical efficacy studies are less meaningful in establishing biosimilarity. Every study conducted as a noninferiority study needs an acceptance criterion before the study is initiated, a parameter that is strictly arbitrary and supposed to be drawn from clinical judgment. Given that there are few data on the long-term use of biosimilars, such conclusions can be only arbitrary, at best.
Second, because biosimilars receive extrapolation of indications, does a single study suffice to remove all residual uncertainty for all indications? The correct answer can only be no. The dependence on clinical efficacy testing has the risk that other elements of dissimilarity, such as may emerge from analytical or clinical pharmacology, will be ignored, leading to the possibility that relying on efficacy studies may result in the approval of biosimilars that are not safe and effective.
Third, since none of the clinical efficacy testing failed, it means that either all products were clinically similar or the protocols were not sensitive enough to differentiate the products. In both cases, the utility of clinical efficacy testing can be questioned. A greater concern arises where a limited clinical efficacy study is conducted to overcome any residual uncertainty in analytical, animal, and clinical pharmacology studies, where the product may be approved in multiple indications with greater risk for safety and efficacy.
In the case of monoclonal antibodies, the mechanism of action is generally known, allowing use of in-vitro/ex-vitro methods to compare efficacy. These tests can be more robust and reliable than the study of clinical responses in patients, particularly in the case of anticancer drugs, where it is almost impossible to secure a treatment-naïve patient population or even a sufficiently large number of patients and there is high variability of response. Do these studies bring added confidence to the safety and efficacy of the biosimilar product?
The issue of immunogenicity testing also needs to be reevaluated for 2 reasons: First, if a product is highly immunogenic, testing it in naïve subjects raises ethical issues; second, if immunogenicity does not alter the pharmacokinetic (PK) profile, then it should not be a major concern, in line with the position taken by the FDA in allowing the waiver of clinical immunogenicity testing of insulins.
The animal pharmacology studies were another area of much confusion and abuse by the biosimilar developers; the FDA had refused to consider multiple safety studies in the evaluation of a product when the FDA rejected one-third of studies as redundant. Typical examples were testing antibodies in species that do not have a receptor to demonstrate toxicity, evaluating immunogenicity in animal species, and dosing animals based on human doses or multiples of human doses. To the best of my knowledge, no study on the testing of biosimilars has ever failed in animal testing because it cannot. The best use of animal testing is in comparative PK profiling, as an extension of analytical similarity testing, where a biological system views the structure and responds to its variability. In India, dozens of monoclonal antibodies are approved by testing in rats because mammalian species, such as the monkey, are not allowed to be tested.
Although the analytical assessment forms the basis for establishing biosimilarity, these studies are unnecessarily complicated due to lack of simple understanding that if the FDA agrees with the release specification of the final product, the tested attributes need not be made part of analytical similarity testing; if it is good enough for the patient, it should be good enough for the FDA! This scientific thesis should limit analytical similarity testing to primary, secondary, and tertiary structure testing and other safety-related attributes.
In conclusion, I believe now is the time for the FDA to lead again in revising the guidance, eliminating all animal toxicology studies, and replacing them with larger-species PK studies, allowing the conduct of human PK studies using novel clinical protocols to combine the PK/pharmacodynamic/immunogenicity testing in a single study, and, where possible, avoid these studies if an in-silico approach can provide the confidence of pharmacologic similarity.
The majority of biosimilar products are monoclonal antibodies where highly relevant receptor binding studies can be conducted to demonstrate safety and efficacy. An excellent example supporting this suggestion comes in the evolution of anticancer treatments, where it is impossible to secure a treatment-naïve population, resulting in high variability in response and in many cases inability to complete the studies. The clinical efficacy testing should not be allowed to overcome any residual uncertainty, and all such products should be rejected as biosimilars under the 351(k) approval pathway and switched over to 351(a) if the developers so desire to continue with the development. The FDA has been forthcoming in taking bold steps that are scientifically based, such as removing clinical immunogenicity testing of insulins, because differences in immunogenicity do not affect the pharmacokinetic profile5.




- FDA. Title VII—Improving Access to Innovative Medical Therapies: Subtitle A—Biologics Price Competition and Innovation. https://www.fda.gov/media/78946/download
- FDA. Biosimilars Guidances. Updated June 21, 2019. Accessed July 10, 2020. https://www.fda.gov/vaccines-blood-biologics/general-biologics-guidances/biosimilars-guidances
- Keown A. FDA Withdraws Draft Guidance on Biosimilar Development. BioSpace. July 3, 2018. Accessed July 10, 2020. https://www.biospace.com/article/fda-withdraws-draft-guidance-on-biosimilar-development/
- Niazi SK. A critical analysis of the FDA draft guidance on development of therapeutic protein biosimilar: comparative analytical assessment and other quality-related concerns. Center for Biosimilars®. Published June 4, 2019. Accessed July 10, 2020. https://www.centerforbiosimilars.com/contributor/sarfaraz-niazi/2019/06/a-critical-analysis-of-the-fda-draft-guidance-on-development-of-therapeutic-protein-biosimilars-comparative-analytical-assessment-and-other-qualityrelated-considerations
- BioSpace. FDA allows waiver of clinical trials for insulin biosimilars as recommended in Niazi citizen petition. BioSpace. Published December 3, 2019. Accessed July 10, 2019. https://www.biospace.com/article/releases/fda-allows-waiver-of-clinical-trials-for-insulin-biosimilars-as-recommended-in-niazi-citizen-petition/
- FDA. Zarxio (filgrastim-sndz). Published April 20, 2015. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/125553Orig1s000TOC.cfm
- FDA. Inflectra (infliximab-dyyb) for injection. Updated July 6, 2016. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/125544Orig1s000TOC.cfm
- FDA. Drug approval package: Erelzi (etanercept-szzs). Published October 25, 2016. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761042Orig1_toc.cfm
- FDA. Drug approval package: Amjevita (adalimumab-atto). Published November 9, 2016. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761024_toc.cfm
- FDA. Drug approval package: Renflexis (infliximab-abda). Published December 10, 2018. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761054Orig1s000TOC.cfm
- FDA. Drug approval package: Cyltezo (adalimumab-adbm). Published September 27, 2018. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761058Orig1s000TOC.cfm
- FDA. Drug Approval Package: Mvasi (bevacizumab-awwb). Published October 10, 2018. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761028Orig1s000TOC.cfm
- FDA. Drug Approval Package: Ogivri (trastuzumab-dkst). Published November 29, 2018. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761074Orig1s000TOC.cfm
- FDA. Drug approval package: Ixifi (infliximab-qbtx). Published November 29, 2018. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761072Orig1s000TOC.cfm
- FDA. Drug approval package: Retacrit (epoetin alfa-epbx). Published December 13, 2018. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/125545Orig1s000TOC.cfm
- FDA. Drug approval package: Fulphila. Published October 26, 2018. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761075Orig1s000TOC.cfm
- FDA. Drug approval package: Nivestym (filgrastim-aafi). Published February 21, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761080Orig1s000TOC.cfm
- FDA. Drug approval package: Hyrimoz. Published March 21, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761071Orig1s000TOC.cfm
- FDA. Drug approval package: Truxima (rituximab-abbs). Published February 25, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761088Orig1s000TOC.cfm
- FDA. Drug approval package: Udenyca. Published March 5, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761039Orig1s000TOC.cfm
- FDA. Drug approval package: Herzuma. Published February 7, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761091Orig1s000TOC.cfm
- FDA. Drug approval package: Ontruzant (trastuzumab-dttb). Published March 5, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761100Orig1s000TOC.cfm
- FDA. Drug approval package: Trazimera (trastuzumab-qyyp). Published May 17, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761081Orig1s000TOC.cfm
- FDA. Drug approval package: Eticovo. Published June 18, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761066Orig1s000TOC.cfm
- FDA. Drug approval package: Kanjinti. Published July 18, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761073Orig1s000TOC.cfm
- FDA. Drug approval package: Zirabev. August 14, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761099Orig1s000TOC.cfm
- FDA. Drug approval package: Ruxience. August 9, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761103Orig1s000TOC.cfm
- FDA. Drug approval package: Hadlima. Published September 5, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761059Orig1s000TOC.cfm
- FDA. Drug approval package: Ziextenzo. Published December 27, 2019. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761045Orig1s000TOC.cfm
- FDA. Drug approval package: Abrilada. Published January 28, 2020. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761118Orig1s000TOC.cfm
- FDA. Drug approval package: Avsola. Published January 30, 2020. Accessed July 10, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761086Orig1s000TOC.cfm
Newsletter
Where clinical, regulatory, and economic perspectives converge—sign up for Center for Biosimilars® emails to get expert insights on emerging treatment paradigms, biosimilar policy, and real-world outcomes that shape patient care.









