




Biosimilar mRNA vaccines, approved under 351(k) or a modified 351(a), can fulfill the present and future needs of accessible life-saving vaccines across the globe.
Business Practice
CME/CE
Subscribe
BIOSIMILAR APPROVALS
- Bone Health
- Immunology
- Hematology
- Respiratory
- Dermatology
- Diabetes
- Gastroenterology
- Neurology
- Oncology
- Ophthalmology
- Rare Disease
- Rheumatology
Biosimilar mRNA Vaccines, Part 2: Fast-to-Market Approach!
The road to production of biosimilar messenger RNA (mRNA) vaccines could be very swift compared with the experience for other biologics. With fewer intellectual property restrictions, small manufacturers could step right in, according to Sarfaraz K. Niazi, PhD.
In my first article on the topic, I created a new term, “biosimilar mRNA vaccine,” to suggest that chemically synthesized messenger RNA (mRNA) vaccines can be approved as biosimilars under the 351(k) provision by the Center for Drug Evaluation and Research (CDER) rather than as standalone or “novel” 351(a) drugs under the Center for Biologics Evaluation and Research (CBER).
However, there are pros and cons to both pathways to approval. In the biosimilar pathway, a 12-year exclusivity protection will apply if the reference vaccine is revised to address new mutations. This would require biosimilar developers to file a new biologics license application (BLA).
The analytical similarity testing can also be challenging depending on which is considered the active ingredient: the mRNA or the translated protein. In a standalone approval, complete testing will take a longer time and cost. However, given the recent guideline on COVID-19–related products, the FDA is willing to consider standalone filing with the same concessions as are allowed in the filing of biosimilar products. It remains to be seen how the first product filing will be treated.
Today, the world needs about 8 billion doses to create herd immunity worldwide; the WHO and other global agencies are urging that vaccines be supplied to countries that could not afford the cost of the first wave of COVID-19 vaccines. Although there is a surplus of vaccines in the United States, 75% of the world’s population is still waiting for shots. To meet this challenge, we will have to develop a novel pathway to secure faster entry into the market for the COVID-19 mRNA vaccine.
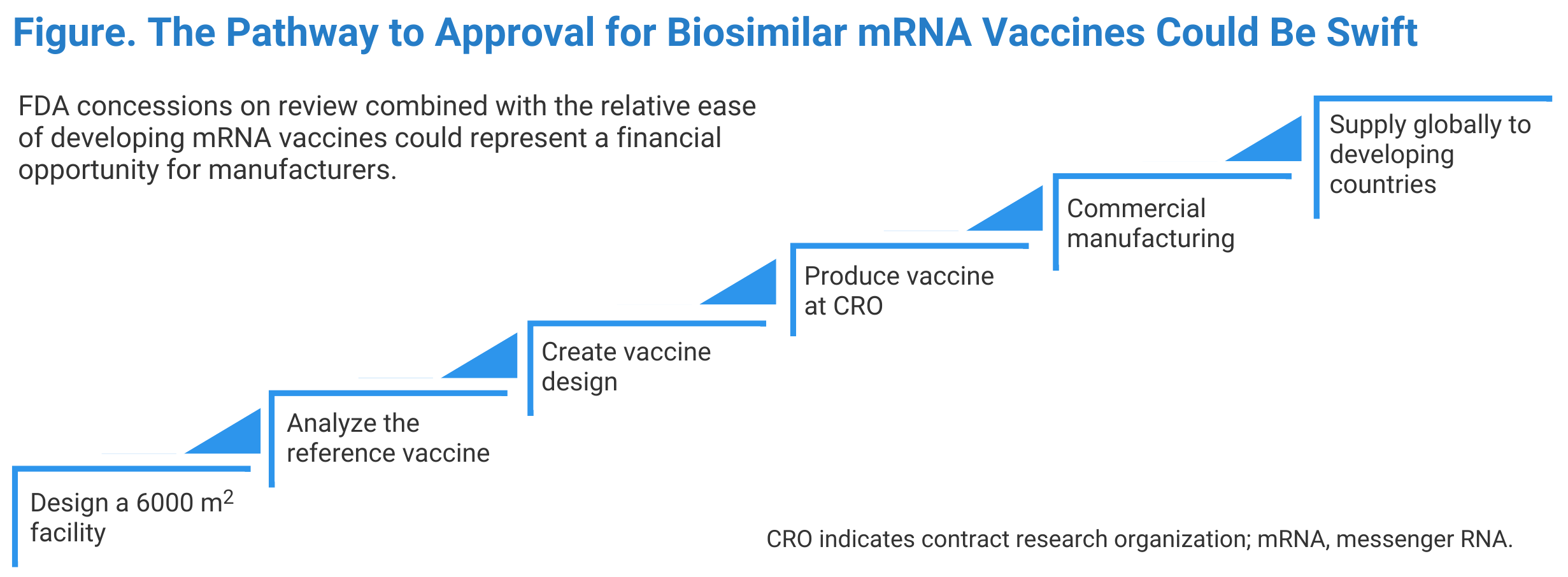
In this article, I present a path that manufacturers could follow for quick access to the market (Figure). To make the best use of the available technology, a creative and well-organized approach is required:
- The chemical synthesis of mRNA is much simpler than manufacturing antibodies and other biological products. Therefore, the production facility setup is a lower-cost step. Additionally, since the vaccine dose is small (micrograms), we need to manufacture smaller quantities (eg, 30 kg to make 1 billion Pfizer vaccine doses). Therefore, an adequate, 6000-square-foot facility can quickly be installed using modular systems within 3 months.
- The only biological step in the production is the creation of DNA from a genetically modified Escherichia coli; thus, this step is not rigorous. A 30 L bioreactor can easily produce 250 million doses per year. Comparing this with production of an antibody such as bevacizumab, a similar number of doses would require 250,000 kg of drug substance. If this antibody process produces 5 g/L of culture media, it would take a 100,000 L bioreactor to produce 5 kg of vaccine, and it would take 5000 batches to produce the required quantity.
- Whereas a certain minimum number of reference product samples are required for testing, there are some ethical constraints in using a commercial vaccine in short supply for research purposes; however, enough sample material for testing could be obtained by pooling vaccine from used vials.
- With a vaccine design that duplicates the reference vaccine, a contract research organization could be employed to manufacture an initial product sample.
- The mRNA vaccine manufacturing process, as with other biological drugs, is heavily patent protected. The onus of proving infringement lies with the claimant. Most significantly, Moderna owns several mRNA-vaccine patents and has allowed their use without claiming infringement. So, a copy of the Moderna vaccine should not be subject to any intellectual property (IP) infringement, notwithstanding any IP Moderna is using under a license.
- By now, this would be the third month of planning, and the facility would be outfitted. After that, the manufacturer would be scheduling the first meeting with the FDA to discuss the regulatory review plan, including analytical, in vitro, and animal model testing for antibody production.
- The sooner a manufacturer decides on the scale of production, the sooner cost estimates can be prepared. For example, producing 1 billion doses of a COVID-19 vaccine will require about 31 kg of mRNA, and the cost will be about $0.68 per dose in a bulk form and about $1 in finished form. About 68% of the cost is associated with the chemicals required to make mRNA, and the rest is packaging. These costs will be lower if the vaccine is manufactured outside the United States where the labor costs are lower. At this point, roughly 6 months would have passed, and it would be time to file the BLA if the intention is to secure FDA approval; otherwise, the unpackaged vaccine will be ready for export. FDA approval is not needed to export in bulk if the vaccine is not in package form for administration.
- Distributing and selling the COVID-19 vaccine requires collaboration with global health agencies, like the WHO. Many countries will prefer to receive the vaccine in bulk to package it, to save cost. Although FDA approval is not required for bulk export, Good Manufacturing Practice compliance is required. In my opinion, everything will go faster if a manufacturer seeks FDA approval. There are many global concerns interested in developing mRNA vaccines that are now providing many funding resources, making this a lucrative business opportunity.
Biosimilar mRNA vaccines, approved under 351(k) or a modified 351(a), can fulfill the present and future needs of accessible life-saving vaccines across the globe. The original developers are not able to offer these products at an affordable cost for many reasons, including their capital investment and their need to show a robust profit because they are publicly traded companies. Since the dose of the vaccine is minimal, the production costs are also low. If the cost of an amount provided as bulk is about $0.68 and sold at $3 per dose, it can generate an income of billions of dollars. Still, the reference vaccine producer will not enter these markets, or else they will lose their most profitable business, supplying to richer countries at almost 10 times the price they would get from developing country patients.
Most people in the poorest countries will need to wait another 2 years before being vaccinated against COVID-19. Around 11 billion doses are required to fully vaccinate 70% of the world’s population against COVID-19. However, the most effective mRNA vaccines are not going to be available to developing countries. Pfizer has cautioned its investors that the rewards of producing a vaccine for low-income and mid-income countries will be nonexistent to very low. The position taken by Pfizer is in stark contrast to Moderna, which has allowed the use of its pending intellectual property (patents). However, with intervention from the US government and the World Trade Organization, Pfizer will allow the use of the IPs if the intent is to distribute these vaccines to the developing countries.
Therefore, I am encouraging companies to consider this remarkable opportunity to help in ending this pandemic and saving millions of lives.
Newsletter
Where clinical, regulatory, and economic perspectives converge—sign up for Center for Biosimilars® emails to get expert insights on emerging treatment paradigms, biosimilar policy, and real-world outcomes that shape patient care.
Recent Videos



Related Content






