

- Bone Health
- Immunology
- Hematology
- Respiratory
- Dermatology
- Diabetes
- Gastroenterology
- Neurology
- Oncology
- Ophthalmology
- Rare Disease
- Rheumatology
Biosimilarity: A Novel Approach to Develop Therapies and Vaccines for COVID-19
The coronavirus disease 2019 (COVID-19) pandemic has created a rush to develop diagnostic, treatment, and prevention modalities that is unique in the past 100 years. As of the end of August, there were more than 3000 studies underway in 115 countries and in every state in the United States, where there were 685 studies in progress. US federal funding had gone to 79 studies in 23 countries. There were 108 vaccines trials underway. To manage this process effectively, the FDA has developed the Coronavirus Treatment Acceleration Program (CTAP), and this has reported more than 570 potential COVID-19 treatment development plans. So far, the FDA has reviewed more than 270 trials and authorized emergency use of dexamethasone and remdesivir; no other COVID-19 treatment approvals are imminent (Figure).


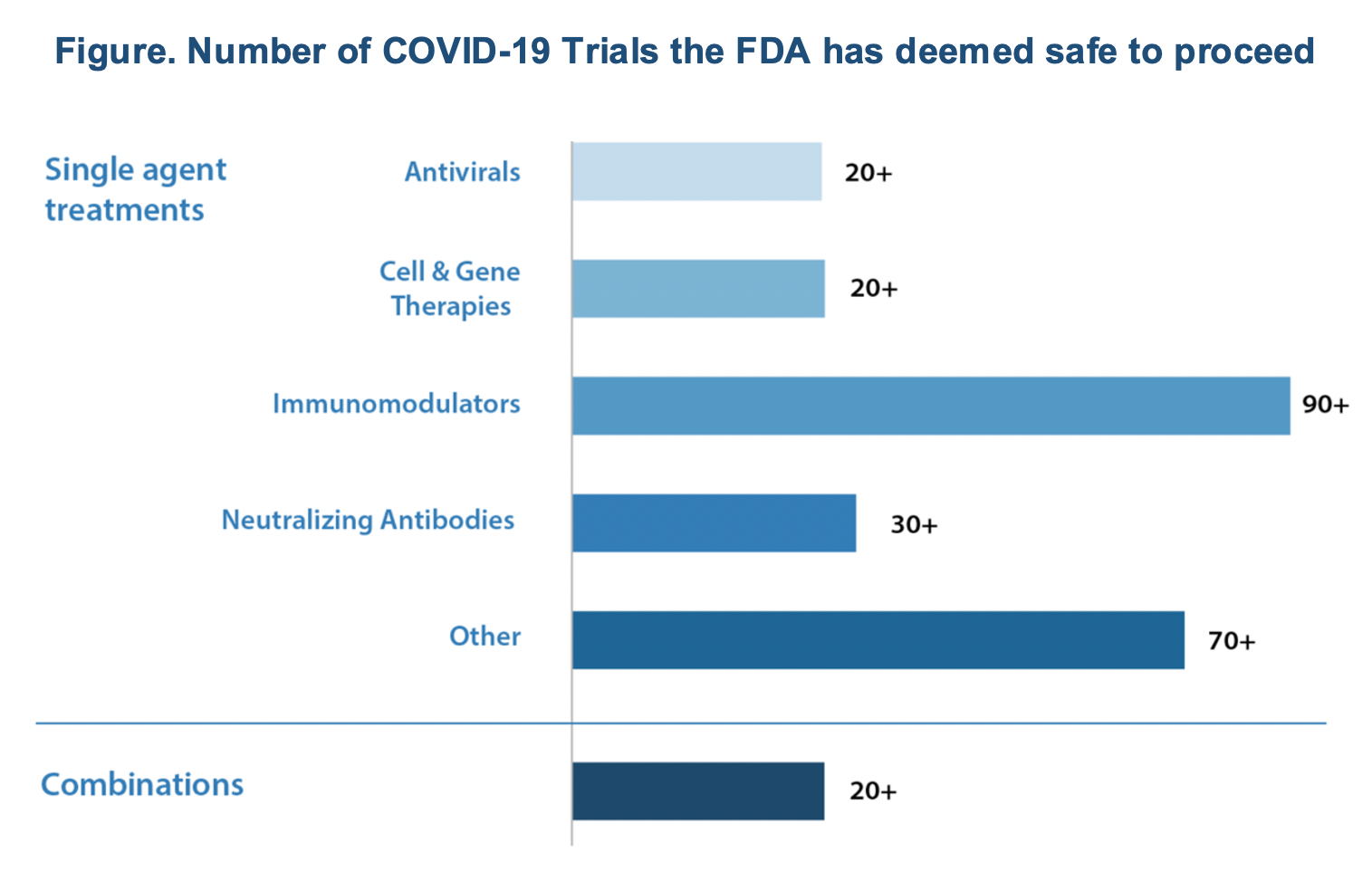
By far, immunomodulators are the dominant type of drugs under investigation as possible treatments for COVID-19 and related conditions.
The diversity of products developed is essential if we are to find a quick solution to controlling the COVID-19 pandemic. The categories of treatment include:
- Antiviral drugs, which keep viruses from multiplying and are used to treat many viral infections (such as HIV, herpes, hepatitis C, and influenza)
- Immunomodulators aimed at tamping down the body’s immune reaction to the virus, such as when the patient’s body overreacts and starts attacking its organs
- Neutralizing antibodies, which may help individuals fight the virus and include manufactured antibodies, animal-sourced antibody therapies, and blood-derived products such as convalescent plasma and hyperimmune globulin, which contain antibodies taken from individuals who have previously had COVID-19
- Cell therapies, including cellular immunotherapies; autologous and allogeneic cells, such as stem cells; and related products
- Gene therapies intended to modify or manipulate the expression of a gene or to alter the biological properties of living cells for therapeutic use. This diversity of products developed is essential if we are to find a quick solution to controlling the COVID-19 pandemic.
The development of vaccines is challenging because they typically require years of research and testing before they reach the clinic. The work to create COVID-19 vaccines began in January 2020 with the deciphering of the SARS-CoV-2 genome. As of this writing, China has approved a Sinovac vaccine for limited use (August 31), and Russia’s Gamaleya has moved to phase 3 testing of a vaccine called Gam-Covid-Vac Lyo, a combination of adenoviruses engineered with a coronavirus gene. The vaccine, dubbed Sputnik V, has conditional approval in Russia pending results of the phase 3 testing. There are no data available to evaluate the safety and efficacy of either of these vaccines.
In June 2020, the FDA issued detailed guidance on the development of COVID-19 vaccines, requiring that a vaccine protect at least 50% of the recipients.
The guidance mandates testing a new vaccine first on cells and then in animals, such as mice or monkeys, to see if it produces an immune response resulting in the production of virus-specific antibodies. In phase 1 trials, the vaccine is given to a small number of healthy volunteers to test safety and dosage as well as to confirm that it stimulates the immune system. In phase 2, the trial is expanded to hundreds of volunteers split into groups, such as children and the elderly, to see if the vaccine acts differently in them. These trials further test the vaccine’s safety and ability to stimulate the immune system. In phase 3 trials, thousands of volunteers receive the vaccine, and investigators wait to see how many become infected compared with volunteers who receive a placebo. These trials can determine if the vaccine protects against the coronavirus.
On August 30, FDA Commissioner Stephen Hahn, MD, announced that the FDA would allow review and approval of vaccines based on preliminary phase 3 trials data. The rush to develop by investigators and the desire to approve by the regulatory agencies have brought us to the brink of decision-making that can have serious consequences. At the same time, a new drug approved for a disease or condition demonstrates the risk based on the number of patients exposed. However, with a vaccine that is given to billions of healthy people and, in the case of COVID-19, a majority of the world’s population, a large number of subjects can be adversely affected even if the percentage of events is small; such risk does not apply to therapeutic products developed to treat patients because of the limited use only in patients.
COVID-19 is a challenge that requires novel approaches to manage. One such strategy is using the principles of establishing biosimilarity to assess the safety and efficacy of both treatments and vaccines. Biosimilars are macromolecules that include antibodies, and their safety and efficacy are tested first by comparing the biosimilar candidate with the reference product to ensure that there is no clinically meaningful difference between them. The analytical science has advanced well over the past few decades to enable us to compare the primary, secondary, and tertiary structures of proteins of all sizes with a high degree of accuracy.
I have suggested to the FDA that it require a test comparing the structure of antibodies produced in patients with COVID-19 (serving as an equivalent of a reference product) with the antibodies produced using a vaccine (as the proposed biosimilar product). This test can be conducted within a few days of administering the first dose of the vaccine. This testing can also be a preamble to full-blown testing to further reduce the risk to study subjects. For comparison, there is no need to conduct all tests required to establish biosimilarity; only a structural comparison is needed. The testing will also include binding or functionality tests and any other novel testing methods that the developers can propose to assure that the antibodies produced by a vaccine are highly similar to the antibodies produced in patients. Another test for structural similarity can be a pharmacokinetic study in animal species such as monkeys, where 3 to 4 animals can be administered the 2 antibodies to determine if there are any differences in their disposition kinetics profile. There is no need for animal toxicology studies, particularly not in any species that does not have receptors to interact with antibodies, such as rodents.
The biosimilarity approach detailed above can also serve as an effective tool to a developer of mRNA vaccines wherein testing in animals can provide extensive information on potential efficacy and safety of the antibodies produced.
The antibodies produced in response to a vaccine can be different to some extent than the antibodies produced from a live virus infection and yet still be useful in providing immunity; and significant structural differences will point to unforeseen safety issues.
The biosimilarity approach presented above and detailed to the FDA will enable the FDA to make an early decision on the safety of clinical trials and significantly speed up the development of COVID-19 vaccines.
The development of treatments is more likely to be successful if, instead of repurposing drugs, as hundreds of trials are trying to prove is possible, we can produce the same antibody that is found in patients infected by live COVID-19. Additionally, there are other opportunities in the scaffold antibodies or smaller proteins that simulate antibody function and are synthesized.
I am confident that although we have suffered through this pandemic, science will come out as a savior, and we will have many more novel opportunities to control diseases in the future.
Newsletter
Where clinical, regulatory, and economic perspectives converge—sign up for Center for Biosimilars® emails to get expert insights on emerging treatment paradigms, biosimilar policy, and real-world outcomes that shape patient care.









