

- Bone Health
- Immunology
- Hematology
- Respiratory
- Dermatology
- Diabetes
- Gastroenterology
- Neurology
- Oncology
- Ophthalmology
- Rare Disease
- Rheumatology
FDA and Industry Experts Unpack Biosimilar Device Requirements
At the GRx+Biosims 2024 conference, a panel of industry experts and FDA officials discussed evolving device requirements for biosimilars and interchangeable biosimilars, highlighting new approaches to comparative use human factors studies, regulatory challenges, and alternative validation methods.
Panelists explored key insights into biosimilar device requirements, emphasizing risk analysis, human factors studies, and alternative validation approaches to streamline regulatory approval for interchangeable biosimilars, while maintaining patient safety and efficacy, at the GRx+Biosims 2024 conference.


Because most biologic drugs are injectable, companies need to design drug delivery devices to administer their products. However, companies may develop designs for the same type of device that have slight differences to other products of the same molecule. | Image credit: RFBSIP - stock.adobe.com
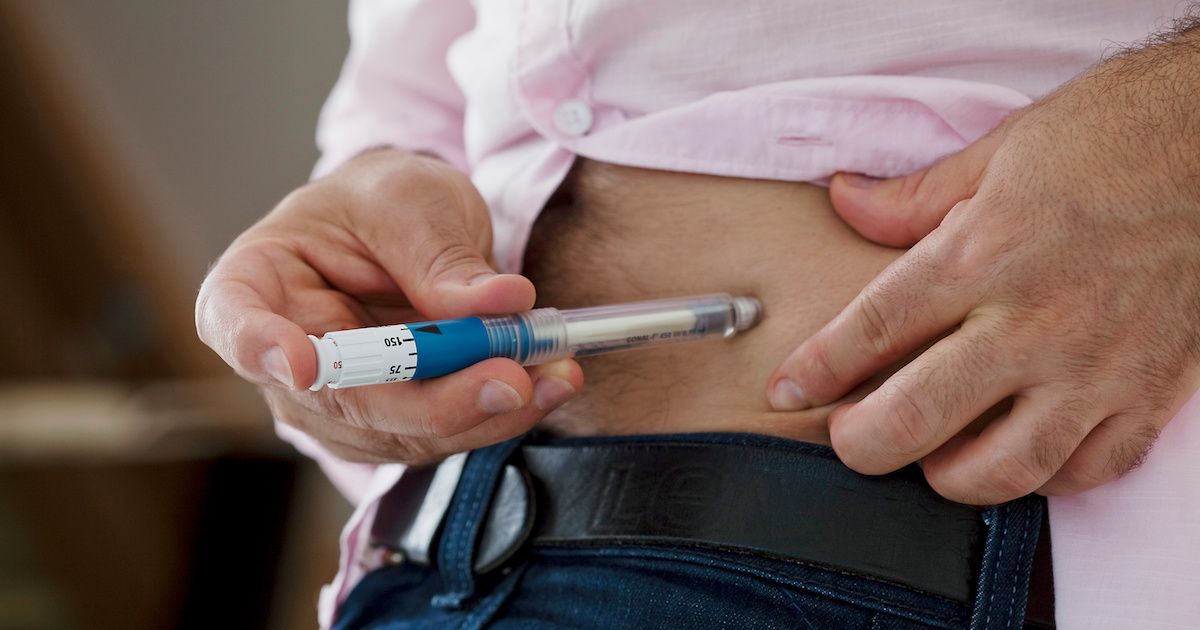
This session focused on providing a comprehensive understanding of the current data and outcomes related to device requirements for biosimilars and interchangeable biosimilars, with insights into how this information might shape future guidance. Additionally, the session offered an overview of the FDA's perspective on interchangeable biosimilars and devices, emphasizing the importance of such guidance in fostering safety and efficacy in biosimilar use. The GRx+Biosims conference was held October 21-23, 2024, in Rockville, Maryland.
Moderator Maria Burkholder, MHA, senior director, regulatory affairs global biosimilars, Teva Pharmaceuticals, began the discussion by defining similarity and interchangeability in the context of generic applications and biosimilars. She explained the distinction between interchangeable and noninterchangeable biosimilars and presented a diagram illustrating the requirements for both to receive approval. The importance of use-related risk analysis and threshold analysis in biosimilar development was emphasized.
Jason Flint, MBA, PMP, deputy director at the FDA’s Division of Medication Error Prevention and Analysis I, clarified that comparative use human factors (CUHF) studies are not the only way to prove substitutability or interchangeability. Cris Ausin, PhD, scientific reviewer for the FDA’s Office of Therapeutic Biosimilars and Biosimilars, added that comparative analyses could be used as alternatives to human factors validation studies.
Johannes Keuschnigg, PhD, regulatory devices portfolio head, Sandoz, outlined the steps for developing an accommodation product, focusing on use-related risk analysis and threshold analysis. He explained the 3 key steps: physical comparison, labeling comparison, and user task comparison. Options for addressing nonminor drug device design differences, such as design changes and CUHF studies, were discussed. He also highlighted the challenges and opportunities in enabling user interface differentiation while maintaining patient safety.
Continuing the discussion, Keuschnigg addressed challenges in planning and executing CUHF studies, including recruitment and cost issues. “If you have 100 patients [in the originator group], you need to give each and every one of those 1 piece of the reference product. We know how much originators can cost. So just the cost of the material for these types of studies gets really significant,” he said.
Keuschnigg suggested potential opportunities, such as dedicated FDA guidance and a risk-based approach. The importance of statistical powering, sample size calculation, and defining noninferiority margins was also emphasized.
Flint discussed alternative validation approaches and the importance of building a strong case for their use. “Typically, what we end up seeing is a [device] difference [between the biosimilar and originator], and [the biosimilar company] goes, ‘Well, we're not worried about it.’ Well, OK, but build the case, right? Tell me the story. Why is [the difference] not wrong? And, and if you're able to do that in the comparative analysis space in a reasonable way, then you're avoiding CUHFs altogether,” he explained.
He mentioned that alternative approaches are being explored, emphasizing the role of comparative analysis in justifying the need for a CUHF study. He highlighted the importance of demonstrating that any device differences do not impact clinical safety or efficacy.
Amith Belavadi, director, technical program management, project and portfolio management at Amneal Pharmaceuticals, presented 2 case studies to illustrate the classification of device design differences. The first involved a simple difference in the delivery mechanism of a device, while the second involved differences in user tasks, comparing a 2-step autoinjector to a 3-step autoinjector. The focus was on determining whether these differences were major or minor and their implications for CUHF studies.
During the Q&A portion, Flint and Ausin emphasized the importance of data and case-by-case evaluations, offering practical tips for submitting protocols and conducting meetings with the FDA. Flint provided updates on the FDA’s progress in streamlining CUHF protocol review and stressed the importance of clear guidance and timelines. The session concluded with a discussion on simplifying requirements and the ongoing need for research and collaboration in this area.
Reference
Ausin C, Belavadi A, Burkholder M, Flint J, Keuschnigg J. Device requirements for biosimilars and interchangeable biosimilars. Presented at: GRx+Biosims; October 21-23, 2024; Rockville, MD.
Newsletter
Where clinical, regulatory, and economic perspectives converge—sign up for Center for Biosimilars® emails to get expert insights on emerging treatment paradigms, biosimilar policy, and real-world outcomes that shape patient care.









