Importance
Wyost/Jubonti is the first biosimilar to reference Amgen's Prolia/Xgeva to receive regulatory approval in the US. They are also the 47th and 48th biosimilars overall to approved by the FDA and ninth and 10th biosimilars to receive interchangeability designations.
For reference, there are currently no denosumab biosimilars approved in the European Union and only 2 approved in Canada.
The FDA has approved Wyost/Jubbonti (denosumab-bddz; GP2411), the first biosimilar to reference Xgeva/Prolia (denosumab) in the US.1 The drugs will be used to treat osteoporosis and hypercalcemia as well as prevent skeletal-related events associated with bone metastases from solid tumors.
The FDA accepted Sandoz’ biologics license application for Wyost/Jubbonti in February 2023. This approval is also notable in that there are currently no denosumab biosimilars approved in the European Union. The news comes after the same products were approved in Canada last month.
Similar to the reference products, the biosimilars have the same drug compound (denosumab-bddz) but were approved with 2 brand names that will be used for different indications:
prevent skeletal-related events (SREs) in patients with multiple myeloma and in patients with bone metastases from solid tumors
postmenopausal women with osteoporosis at high risk for fracture
adults and skeletally mature adolescents with giant cell tumor of bone that is unresectable or where surgical resection is likely to result in severe morbidity
to increase bone mass in men with osteoporosis at high risk for fracture
hypercalcemia of malignancy refractory to bisphosphonate therapy
to treat glucocorticoid-induced osteoporosis in men and women at high risk for fracture
to increase bone mass in men at high risk for fracture receiving androgen deprivation therapy for nonmetastatic prostate cancer
to increase bone mass in women at high risk for fracture receiving adjuvant aromatase inhibitor therapy for breast cancer
____________
Jubbonti is interchangeable with Prolia, and Wyost is interchangeable with Xgeva.
Wyost and Jubbonti were also approved with interchangeability designations, allowing them to be substituted for their respective reference product at the pharmacy level without needing to wait for a provider to give permission. It is the ninth biosimilar to receive this label.
"Sandoz has achieved the first FDA approval for biosimilars to denosumab, a medicine that can address primary and secondary bone loss, such as osteoporosis, as well as cancer-related skeletal events, which are disease states that can profoundly reduce quality of life for patients. I am proud that Sandoz continues to pioneer access to these life-changing medicines for the patients who need them most," commented Keren Haruvi, President Sandoz North America, in a statement.
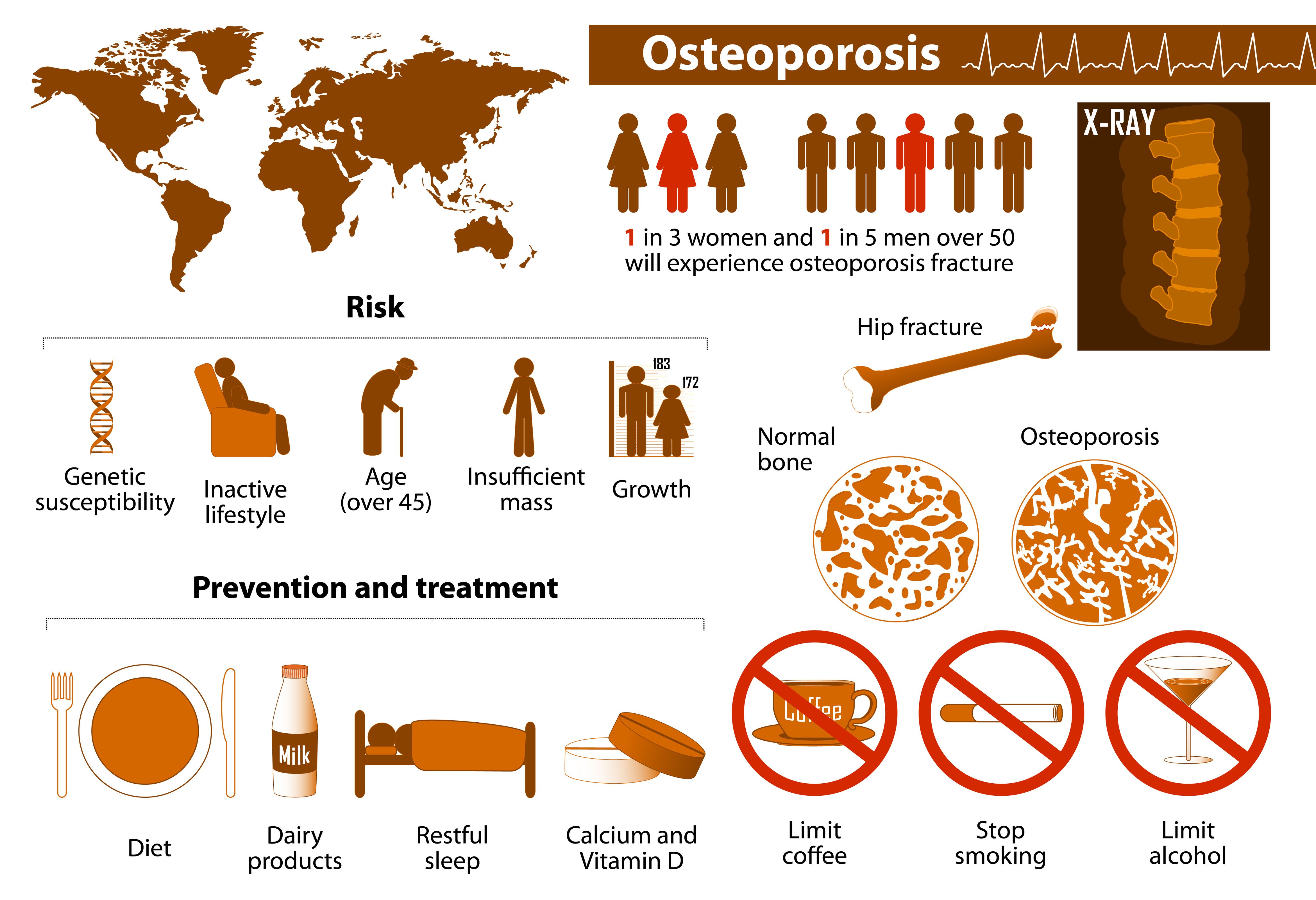
According to Sandoz, over 10 million adults over age 50 in the US have osteoporosis, of whom most (> 80%) are women. It is predicted that 1 in 2 women and 1 in 4 men will experience an osteoporosis-related fracture in their lifetime, which could result in decreased quality of life, disability, and death.
The approval of Wyost/Jubbonti was based on a comprehensive data package containing phase 1 and phase 3 study results that were presented at the American Society of Clinical Oncology annual meeting 2023 and the American Society for Bone and Mineral Research annual meeting 2023, respectively.
Denosumab products are human monoclonal antibodies that bind to the RANKL protein, preventing activation of the RANK receptor on the surface of osteoclasts and preventing osteoclastic bone resorption.
The phase 1 study evaluated the pharmacokinetics (PK) and pharmacodynamics (PD) of denosumab-bddz compared with the reference product (EU authorized and US licensed) in 499 healthy participants (mean [SD] age, 64.6 [6.1] years; White, 90.9%).2 The double-blind, 3-arm, parallel-group analysis was conducted at 2 German centers. Participants were randomized 1:1:1 to receive the biosimilar or the EU- or US-sourced reference agents.
Participants were between 28 and 65 years old and weighed between 50 and 90 kg. PK end points were measured by area under the serum concentration-time curve measured from the time of dosing and extrapolated to infinity (AUCinf), or to the last measurable concentration (AUClast), and maximum observed serum concentration (Cmax). PD end points were measured by the area under the effect-time curve of the percentage change from baseline (AUEC %CfB) in serum carboxy-terminal crosslinked telopeptide of type I collagen (CTX) and CTX and procollagen I N-terminal propeptide (PINP) serum concentration per visit.
Results showed that the treatment arms (biosimilar vs EU-sourced reference product; biosimilar vs US-sourced reference product) showed statistically similar values for AUCinf (1.09; 1.06), AUClast (1.08; 1.05), Cmax (1.02; 1.00), and AUEC %CfB in CTX (1.01; 1.01). Changes from baseline to Week 39 in serum concentrations of CTX and PINP were also similar between groups.
The international, double-blind, parallel group phase 3 trial (ROSALIA study; NCT03974100) looked at the clinical efficacy, safety, and tolerability of denosumab-bddz compared with the reference product through 52 and 78 weeks.3 Postmenopausal women with osteoporosis aged 55 to 80 years who had bone mineral density (BMD) T-scores between 2.5 and 4.0 at the lumbar spine were eligible for participation.
In total, 527 patients were randomized 1:1 to receive a 60-mg dose of Wyost or the EU-authorized reference product on day 1 and at week 26. Prior to the third dose at week 52, patients who were receiving the reference product were rerandomized to remain on the reference product or switch to the biosimilar.
Overall, 502 patients received a third dose (biosimilar to biosimilar, n = 253; reference to reference, n = 125; reference to biosimilar, n = 124). The mean age of the participants was 64.6 (6.15) years, and most (91.3%) were White.
No clinically meaningful differences between the groups regarding new vertebral fractures were observed. Switching from the reference product to the biosimilar did not affect the incidence or severity of treatment-emergent adverse events (TEAEs). Immunogenicity profiles were similar between groups.
Most TEAEs were grade 1 or 2, most of which were not considered treatment related, and no treatment discontinuations occurred as a result. The most common TEAEs reported were hypocalcemia, COVID-19 infection, and nasopharyngitis.
The majority of antidrug antibodies had very low, nonreportable titers (< 20 mg/mL), were nonneutralizing, and transient.
In its company statement, Sandoz declined to comment on a launch timeline due to ongoing litigation issues with the reference product manufacturer (Amgen).
How do you think bone health specialists will react to the availability of denosumab biosimilars?
References
1. Sandoz receives FDA approval for first and only denosumab biosimilars. News release. Sandoz; March 5, 2024. Accessed March 5, 2024. https://www.marketscreener.com/quote/stock/SANDOZ-GROUP-AG-160016887/news/Sandoz-receives-FDA-approval-for-first-and-only-denosumab-biosimilars-46097288/
2. Vogg B, Poetzl J, El Galta R, Fuhr R, Schwebig A, Sekhar S. Pharmacokinetics and pharmacodynamics of proposed denosumab biosimilar and reference denosumab in healthy male subjects. Presented at: 2023 ASCO Annual Meeting; June 2-6, 2023; Chicago, IL. Poster SAT-436.
3. Jeka S, Dokoupilová E, Kivitz A, et al. Pharmacokinetics and pharmacodynamics of proposed denosumab biosimilar and reference denosumab in postmenopausal osteoporosis: the ROSALIA study. Presented at: ASBMR 2023 Annual Meeting; October 13-16, 2023; Vancouver, Canada. Poster P303.