

- Bone Health
- Immunology
- Hematology
- Respiratory
- Dermatology
- Diabetes
- Gastroenterology
- Neurology
- Oncology
- Ophthalmology
- Rare Disease
- Rheumatology
Study: Biosimilar vs Originator Spending on Comparative Efficacy Trials
Investigators evaluated trial spending and reasons for biologics license application failures.
A study of comparative clinical efficacy trials for biosimilars, often considered unnecessary, suggests that these trials are often larger and as rigorous as those done for originator products, and often are more costly than for new molecular entities.
The study evaluated biosimilar trials conducted for products approved between January 2010 and October 2019. “When phase 3 efficacy trials were required (21 of 23 approved biosimilars), the median patient enrollment, treatment duration, and estimated costs often exceeded those reported in published studies of FDA pivotal trials required for new molecular entities,” authors of the study wrote.
Comparative clinical efficacy studies are designed to confirm the clinical equivalence of a biosimilar candidate to its reference product using a study population, prespecified margins, and an efficacy end point. These studies help elucidate the safety and immunogenicity, or potential to generate an immune system response, of biosimilars vs reference products.
Critics of comparative clinical efficacy studies contend that these trials are generally wasteful and do not produce data that are nearly as useful in proving biosimilar equivalence as earlier-stage pharmacokinetic and pharmacodynamic studies. One reason biosimilar sponsors continue to do comparative in-human studies is that they help to convince providers that biosimilars are equivalent to reference products because providers are more accustomed to studying comparative results for their product information.
Findings
Investigators found that the median cost of 29 comparative efficacy studies for biosimilars was $20.98 million. For 24 of these studies, the median enrollment was 538 with an average treatment duration of 55 weeks and a median cost of $27.6 million (Table).


Click to enlarge
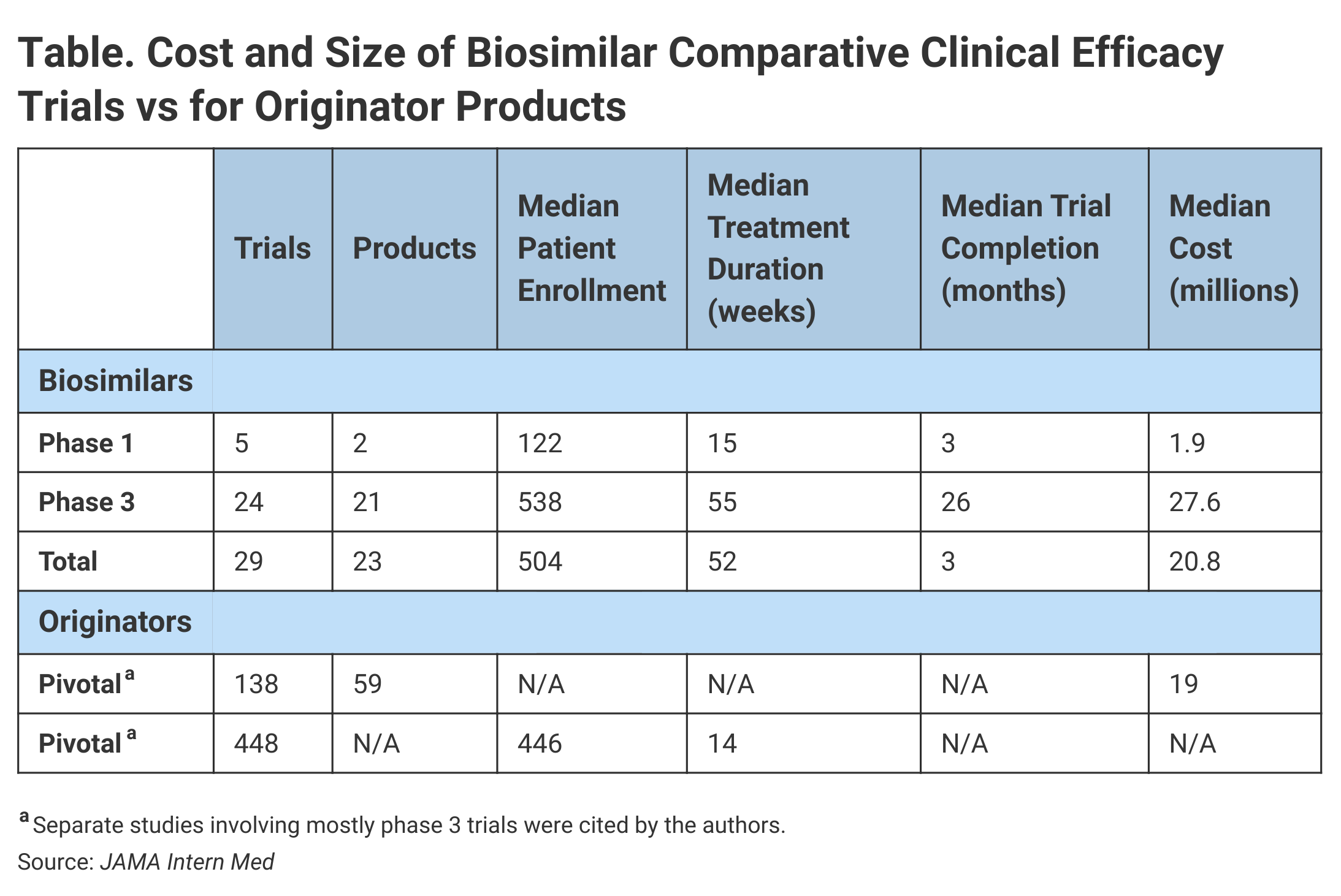
By comparison, the median cost for 138 pivotal trials that supported the approvals of 59 new molecular entities from 2015 to 2016 was $19 million, although investigators added a caveat. “They are not exact comparisons given that the previous findings included biologic products and small-molecule drugs and that phase 3 trials were usually but not always required for marketing approval.”
With regard to enrollment in nonbiosimilar trials leading to FDA approvals from 2005 to 2012, the median patient enrollment was 446 in phase 3 studies.
For the 29 comparative efficacy studies for biosimilars, the median treatment duration was 52 weeks. For 100% of the phase 3 biosimilar efficacy trials, the treatment duration exceeded 26 weeks. Investigators said that just 35% of the pivotal trials supporting approvals in 2015 and 2016 had treatment durations that exceeded 26 weeks.
One characteristic of these trials was that trial duration was generally extended because investigators sought to enroll patients at multiple global sites. The phase 3 studies lasted a median of 26 months from reported start to actual completion, although phase 1 studies were finished in a median of 3 months.
Investigators also looked at FDA reviews of biologics license applications (BLAs) and found that biosimilar reviews took a similar amount of time to complete—a standard 12 months—and none qualified for expedited review. They said 14 biosimilar BLAs completed without delays were reviewed in a mean time of 350 days.
A look at FDA inspections of manufacturing facilities and clinical trial sites showed that the FDA halted BLA reviews for 9 of 23 biosimilars (39%) following these on-site inspections and issued complete response letters stating that the FDA reviews could not proceed until deficiencies noted in the inspections were remedied. Common deficiencies were product quality, manufacturing process, and facility inspection failures.
According to the study, it took biosimilar sponsors from 59 days to 433 days to fix these problems and resubmit their BLAs.
The investigators noted that the global nature of the biosimilar comparative efficacy trials was a challenge for the FDA review process, often requiring on-site inspections. A mean of 12 countries were involved in the trials included in the review.
“The FDA performance and actions in reviewing biosimilar applications were timely and had analytical and inspectional depths that were similar to those seen in previous reviews of new molecular entities,” investigators wrote.
Conclusions
Speculating on why comparative clinical efficacy trials are larger, more costly, and longer than those for new molecular entities, investigators said that when small differences between drugs must been determines, patient enrollment numbers must be large to demonstrate statistical significance of findings. Further, a high level of confidence (CI, 90%-100%) must be established to satisfy FDA requirements for noninferiority trial designs.
Authors of the study concluded that it may be advisable to expand the size and scope of comparative clinical efficacy trials new drug candidates, “given that pivotal trials for new molecular entities provide the primary and essential new scientific evidence that will guide the treatment of thousands to millions of future patients, an ethical and policy question arises about whether numerous, larger, and more costly trials should be conducted to demonstrate that no meaningful differences exist from a proven treatment.”
Reference
Moore TJ, Mouslim MC, Blunt JL, Alexander GC, Shermock KM. Assessment of availability, clinical testing, and US Food and Drug Administration Review of Biosimilar Biologic Basics. JAMA Intern Med. 2021;181(1):52-60. doi:10.1001/jamainternmed.2020.3997
Newsletter
Where clinical, regulatory, and economic perspectives converge—sign up for Center for Biosimilars® emails to get expert insights on emerging treatment paradigms, biosimilar policy, and real-world outcomes that shape patient care.









