

- Bone Health
- Immunology
- Hematology
- Respiratory
- Dermatology
- Diabetes
- Gastroenterology
- Neurology
- Oncology
- Ophthalmology
- Rare Disease
- Rheumatology
The Difference Between an Interchangeable Biosimilar and One That Isn't
The anatomy of an interchangeability switching study.
Permitting automatic substitution of biosimilars for originator brand biologics at the pharmacy counter is often talked about as a potential money saver. Some integrated health care networks already direct pharmacists to intervene on prescriptions to ensure that biosimilars are substituted for higher-cost originator agents. But although the FDA has approved 29 biosimilars, none have interchangeable designations.

That appears likely to change soon. Boehringer Ingelheim recently expressed confidence that results from its phase 3 VOLTAIRE-3 study of the adalimumab biosimilar Cyltezo (BI695501) meet the FDA’s criteria for an interchangeable biologic designation. Cyltezo was approved as a biosimilar in 2017, but owing to the FDA’s unique approval system for biologics, it must pass through a different set of hoops to qualify for interchangeable status.
Similarly, Viatris, a company recently formed by the merger of Mylan with Pfizer’s Upjohn division, is seeking FDA approval for biosimilar and interchangeable designations for the insulin glargine product Semglee. This agent launched in August 2020 in the United States, just not as a biosimilar. It is considered a biosimilar in the European Union, however, where it has been available since March 2018.
So, what does it take for a biosimilar to be approved as interchangeable in the United States? To have its adalimumab biosimilar considered interchangeable to just the 40-mg/0.8-mL formulation of the reference product (Humira), Boehringer Ingelheim had to do a separate study that involved patients switching multiple times from reference drug to biosimilar and back again (Chart).


Click to enlarge.
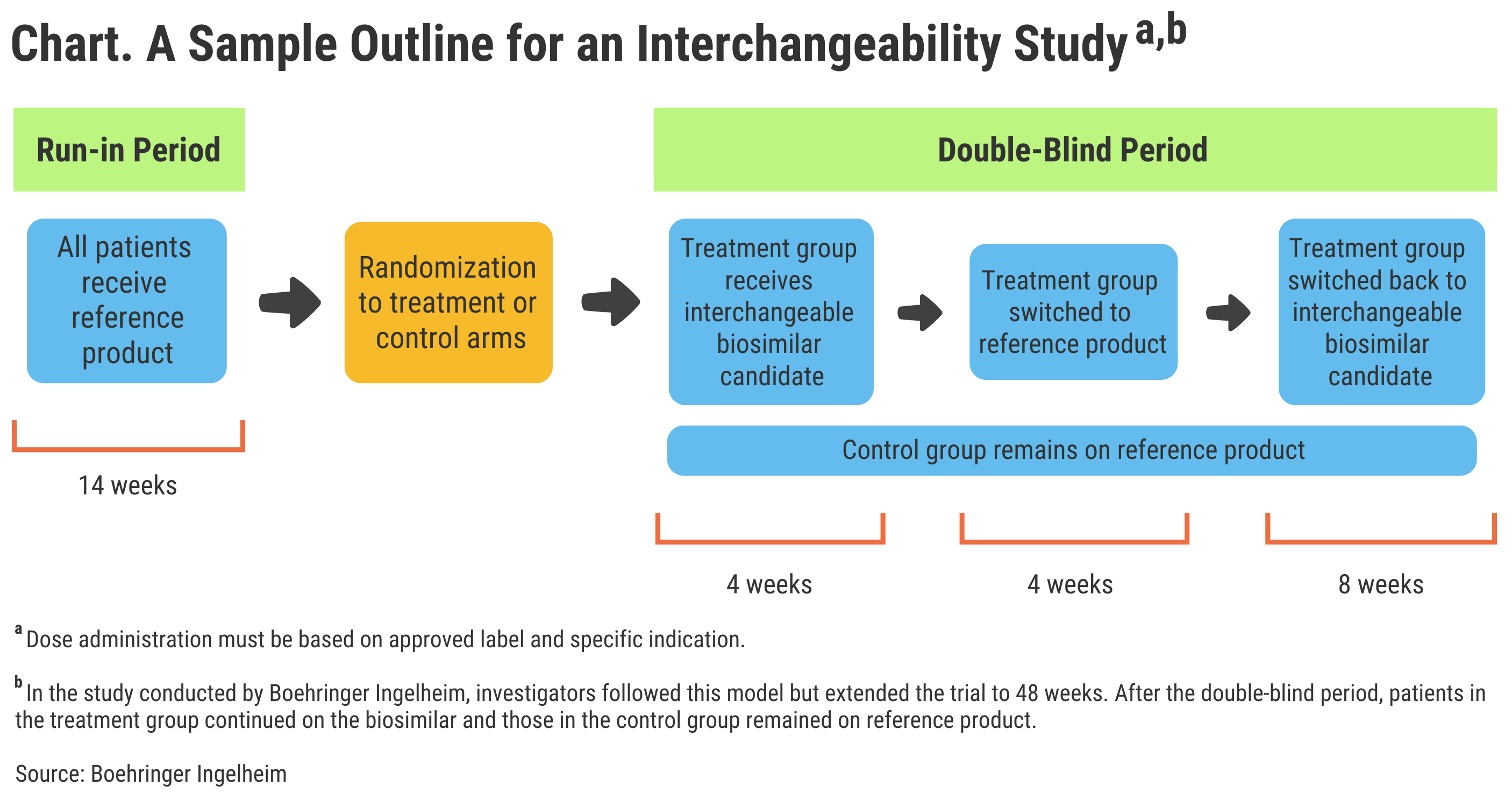
A Separate Regulatory Designation
“An interchangeable biosimilar is a separate additional regulatory designation on top of the high FDA standards for a biosimilar,” Thomas Seck, senior vice president of Medicine and Regulatory Affairs at Boehringer Ingelheim, told The Center for Biosimilars®.
“In addition to meeting the requirements of biosimilarity, an interchangeable biosimilar must first have a highly similar profile and the same clinical result as the reference product in any given patient. Then it must additionally demonstrate that the risk in terms of safety or diminished efficacy of switching with the reference product is not greater than staying on the reference product,” he said.
Interchangeability studies that switch patients between reference product and biosimilar multiple times can help elucidate whether outcomes will be different for a given biosimilar compared with the reference product. These studies can build up physician confidence that patients who are switched to the biosimilar will not be subjected to different pharmacokinetics, immunogenicity, safety, or efficacy than they would if they remained on the reference product.
Interchangeability designations from the FDA would not override state laws on biosimilar substitution. In fact, many states have passed legislation protecting a physician’s right to retain authority over biosimilar use. Some biosimilar proponents might argue that there should be no opposition to automatic substitution following an FDA decision that a biosimilar is as safe and efficacious as a reference product with no clinically meaningful differences, coupled with a switching study to back up interchangeability.
However, state legislatures often see things differently.
“Regulating biologics raises new issues for both state and federal policymakers. Because of their complexity, biologic drugs are much more difficult to replicate than the chemically produced generics for other drugs. The cell lines used and modifications in the manufacturing process affect biologic medicines. As a result, truly identical [biosimilar] versions are currently virtually impossible to produce,” the National Conference of State Legislatures stated in 2019. “Once patents expire for the existing brand name biologic drugs, biosimilar medicines can be produced, which is an occurrence that raises regulatory issues in the states.”
State Management of Interchangeability
An example of this hesitancy to unleash biosimilars at the state level can be found in Connecticut, where legislators recently attempted to amend the law to require private payers and Medicaid to cover originator biologics, not biosimilars, unless providers specify in writing or by electronic means that biosimilars are to be used.
In the European Union, the European Medicines Agency leaves it up to member countries whether to allow automatic substitution of biosimilars. In France, pharmacy level substitution of biosimilars for reference products has long been encouraged and permitted in cases where the physician has not expressly directed otherwise.
For Boehringer Ingelheim, the standards for biosimilar regulatory approval and, on top of that, interchangeability, are a point of contention. The pharmaceutical company hopes to get the FDA to agree that clinically inactive ingredients included in a biologic formulation should not be a factor in whether an approved biosimilar can be used in different concentrations.
“Under the FDA’s interpretation of ‘strength,’ any currently approved [lower-concentration] adalimumab biosimilar (including Cyltezo) cannot be considered biosimilar or interchangeable to the same dose of Humira’s high-concentration formulation,” according to Seck. “This interpretation affects all the currently approved biosimilar versions of Humira and has the potential for wider implications to other biologics and biosimilar and/or interchangeable competition.”
The company has filed a citizen’s petition with the FDA appealing for a change in the interpretation of strength of formulation. A wording change in the company’s favor would be a significant win for the biosimilars community because AbbVie, the maker of the originator adalimumab product (Humira), has captured over 60% of the market for adalimumab in the European Union with its high-concentration, 100-mg/mL dosage form, according to IQVIA. In the United States, the respective percentage for high concentration adalimumab is 70%. And when it comes to displacement of just the 40 mg concentration formulation of adalimumab, the high concentration takeover has been estimated at 90%.
Yuflyma Becomes First High Concentration Challenger
Celltrion Healthcare, in February 2021, received European Commission approval to market a similar concentration biosimilar version (Yuflyma), but that was the first competitor product to gain access to the high-concentration market. Humira was first approved in a 40-mg/0.8-mL strength in 2002, and the FDA approved additional strengths and concentrations over the years. The higher-concentration formulation was first marketed in July 2018.
“We continue to work closely with the FDA, industry peers, payers, health care providers, and patient advocates, engaging in a transparent and public discussion of the issues we have raised, and Boehringer Ingelheim believes that correction of the interpretation of ‘strength’ by the FDA may increase access to more affordable biosimilar and interchangeable biological products approved via the 351(k) pathway,” Seck told The Center for Biosimilars®.
For more about Cyltezo, click here.
Newsletter
Where clinical, regulatory, and economic perspectives converge—sign up for Center for Biosimilars® emails to get expert insights on emerging treatment paradigms, biosimilar policy, and real-world outcomes that shape patient care.









